Explorando cómo son las proteínas por dentro y cómo se mueven: historia, actualidades y retos del grupo de Bioquímica Estructural del IBt
Enrique Rudiño Piñera
Palabras clave: alosterismo, biología estructural, rayos X, cristalografía de proteínas, enzimas
En búsqueda de una pasión
Soy parte de la primera generación en mi familia que tuvo la oportunidad, y el apoyo, para llegar a la universidad, y eso, siempre me llenará de orgullo. Pero también debo confesar que lo que pasó después me llenó de dudas por períodos largos. En particular, cuando terminé mi licenciatura me quedó claro que no había encontrado un tema que me apasionara, uno de esos que me dejara la tranquilidad emocional de dedicarme a él durante el resto de mi vida profesional.
Ese tema apareció durante mis estudios de posgrado y se llama biología estructural: esa área de la ciencia moderna que permite amalgamar a la física, las matemáticas, la computación, la química y, por supuesto a la biología, de una manera que a mis ojos es simplemente poética. Y cuando se hablaba de biología estructural en la época que yo estaba iniciando mis estudios doctorales, eso quería decir que debías emprender el —a veces tortuoso— camino de comprender y dominar a la cristalografía de proteínas utilizando técnicas de difracción de rayos X para descifrar la estructura interna que se logra en un cristal; el arreglo espacial de las cadenas polipeptídicas, conocer y localizar los elementos de organización que hay entre varias copias de proteínas similares (oligómeros), saber dónde hay una estructura llamada alfa-hélice, y donde otra conocida como una hebra beta, etc. [Fig. 1]. Lo que seguía en mi carrera era claro: ahora que había encontrado mi pasión debía hallar donde especializarme.
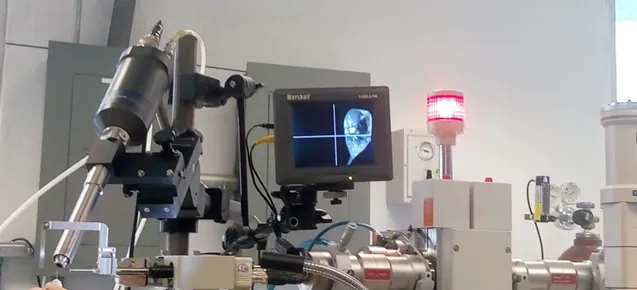
Figura 1. Equipo de difracción de rayos X localizado en el Instituto de Química de la UNAM en la Ciudad Universitaria de la CdMx. En la figura se ven los diversos equipos que permiten posicionar, preservar y colectar datos de difracción, que posteriormente darán lugar a una estructura tridimensional. [Foto: MC. Angela Escudero García].
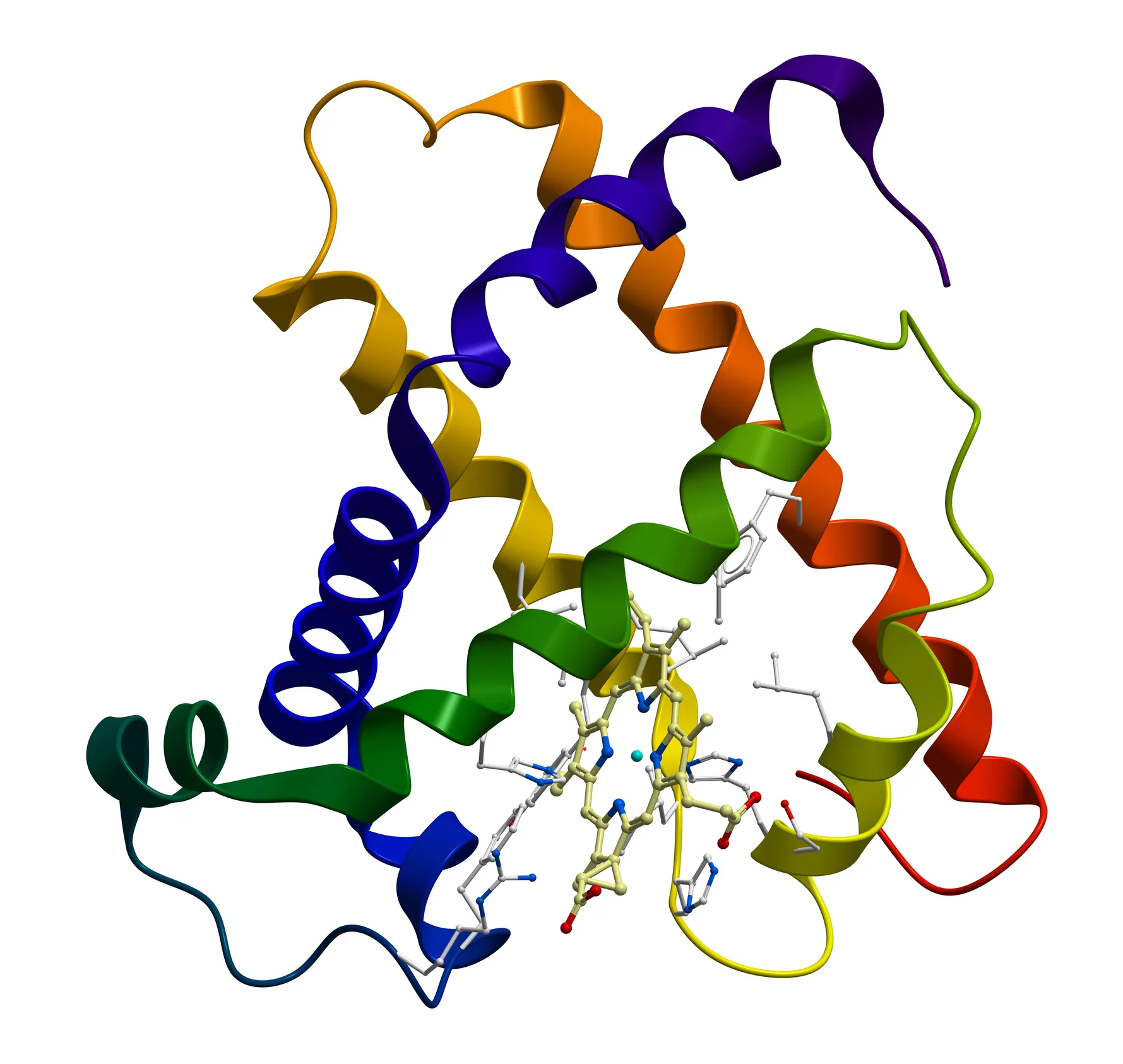
Ya aceptado en un programa de doctorado en Dinamarca, me enteré de que había un grupo de investigación en el Instituto de Biotecnología, en el campus Morelos de la UNAM, que trabajaba con un tipo de proteínas llamadas ‘alostéricas’ (por la naturaleza de otros factores y sitios en la molécula, que regulan su actividad). Este grupo lo dirigía el Dr. Eduardo Horjales Reboredo; un personaje entrañable y de trato rudo pero que me compartió su pasión: tratar de entender mediante estudios de cristalografía —el análisis de moléculas idénticas, apiladas ordenadamente en una estructura que llamamos cristal—, el comportamiento de una enzima alostérica. Lo novedoso no era trabajar con enzimas alostéricas, ya que estas habían sido centro de muchas investigaciones y desarrollos tanto teóricos como prácticos, desde principios de los años 60 del siglo XX; desarrollos y descubrimientos que le permitieron a Jacques Monod ser laureado con el premio Nobel de Medicina en 1965. Lo novedoso estaba en usar a la cristalografía para comprender “visualmente”, como ocurrían los fenómenos alostéricos. Es decir, el interés por estas enzimas alostéricas radica en que tienen una especie de ‘botón de encendido y apagado’ para cambiar de un estado activo (estado R) a otro inactivo (estado T). Dentro de este grupo de enzimas teníamos acceso a estudiar la glucosamina 6-fosfato desaminasa, (abrev. GlcN6PDea), de Escherichia coli.
Una entrevista decisiva
Conseguí una cita con el Dr. Horjales y por más de dos horas me habló del proyecto que me ofrecía para hacer el doctorado bajo su tutoría y de diversos detalles aun sin resolver, para convencerme de tomar el proyecto. Habló de las teorías que pretenden explicar cómo transitan las enzimas alostéricas de un estado activo (R) a uno inactivo (T) y de lo que se necesita para poder cristalizar a una proteína: toda una colección de protocolos y consideraciones particulares que implica, organización, talento, suerte y paciencia al más puro estilo budista [ver Figs. 3 y 4]. Explicó cómo y en donde bombardear a estos cristales directamente con rayos X para difractarlos (desviarlos de una manera específica al rozar o rebotar con las nubes de electrones que rodean a los átomos que forman al cristal, siguiendo reglas develadas hace más de 100 años por una dupla padre-hijo de apellido Bragg). De cómo obtener una familia de puntos en un detector que reflejen —dependiendo del ángulo, tiempo de exposición, el orden en el cristal y mucho análisis utilizando cálculos computacionales— la estructura tridimensional de proteína cristalizada.
En esa misma ocasión, me mostró especialmente, las primeras representaciones tridimensionales de proteínas [Fig. 2], recién calculadas, en una computadora Silicon Graphics utilizando lentes estereoscópicos. Estas imágenes me dejaron mudo de la impresión: estaba viendo en el monitor de la máquina, una red en volumen, que representaba a las nubes de electrones, de cada átomo y de cada aminoácido de una proteína alostérica. No solo me impactó escuchar sobre todos los temas que manejaba, también me asombró la pasión con la que lo relataba y, sobre todo la sensación de ver detalles o implicaciones que nuestros sentidos no pueden percibir directamente, sino a través del conocimiento científico y de sus desarrollos tecnológicos. Y dije adiós a mi plan en Dinamarca y di la bienvenida a la que ha sido mi casa por poco más de 26 años, el Instituto de Biotecnología de la UNAM.
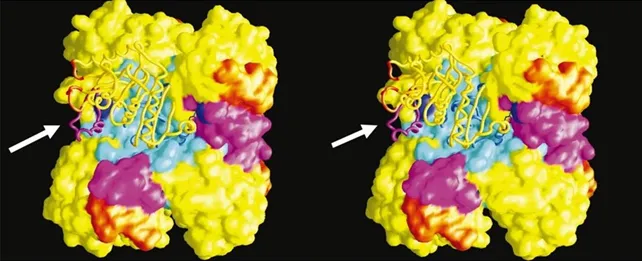
Figura 2. Representación estereoscópica de la superficie de una proteína alostérica: la glucosamina 6-fosfato desaminasa de E. coli. Imagen utilizada en las presentaciones ante su comité tutoral del entonces M. en C. Enrique Rudiño Piñera (1999).
Los inicios del grupo de cristalografía de proteínas del IBt
Eduardo Horjales dirigía un grupo pequeño y compacto en intereses y temas; el alosterismo enzimático era el pretexto, su explicación a nivel estructural era el centro de nuestro quehacer, y las correspondientes glucosaminas 6-fosfato desaminasas de distintos organismos, nuestro modelo experimental. A Eduardo se deben los primeros viajes en 1998 para acceder a “fuentes de radiación de alta brillantez” también conocidas como sincrotrones; bajo su tutoría -en un lapso de 4 años- mi tesis de investigación doctoral estaba lista y mi futuro se movía hacia realizar una estancia posdoctoral fuera de México. El inicio de ese posdoctorado se dilató de más, ya que tuve el honor de ser contratado por el mismo grupo del Dr. Horjales, pasando de una semana a otra, de alumno de doctorado (a veces llamados doctorantes), a investigador asociado: la categoría “de arranque” para seguir formándose en un área de investigación científica.
Años después tuve el privilegio de ser aceptado en el grupo de la Dra. Elspeth F. Garman en la Universidad de Oxford en el Reino Unido, para hacer el tan ansiado posdoctorado, un período de investigación que me abrió los ojos y me diversificó aún más en mis pasiones e intereses. El Dr. Rudiño que regresó al IBt tenía objetivos ahora centrados, no solo en la estructura de las proteínas, sino en cómo se mueven éstas; en los métodos para convertir un patrón de difracción en un modelo tridimensional y, sobre todo, en el efecto y posibles aplicaciones de los daños causados por los rayos X al interactuar con los cristales de proteínas. Esos son los temas que, en nuestro laboratorio, hemos continuado desarrollando una vez que, como Líder Académico, me hice cargo del grupo en 2009 transformándolo en el “Grupo de Bioquímica Estructural” del IBt. Este cambio de nombre enfatiza el hecho de que migrábamos, de una pequeña comunidad académica basada en una técnica experimental, a una que pretendía explicar mecanismos enzimáticos ‘clásicos’, los de tipo alostérico y de movilidad molecular (ya que las enzimas vibran, se tuercen un poco y tienen espacios de tránsito para iones, metabolitos y otras moléculas). Estos mecanismos conformarían una gama más amplia de modelos de estudio, donde el foco iba migrando de las enzimas alostéricas a las metalo-proteínas (aquellas que requieren átomos de hierro, zinc u otros metales para modificar a sus sustratos y transformarlos en productos). Y como suele ocurrir, las preguntas más interesantes son las básicas de inicio: ¿cómo ocurre un proceso enzimático?, ¿cómo se encuentran las moléculas reactivas en la orientación correcta? ¿cómo se disminuye la energía necesaria para que las enzimas transformen a sus sustratos? Y también ¿cómo las puedo manipular (mejorándolas o… en muchas ocasiones, empeorándolas)? y aún más ¿cómo esta información nos permite comprender a los procesos que definen a lo que denominamos como “vida”? …aunque siempre desde la mirilla poderosa pero incompleta de los datos a nivel molecular.
Sobre cómo transformar un problema en una herramienta
Cuando se considera que la gran mayoría de la información tridimensional de proteínas que disponemos —la cual está depositada en el acervo digital del [Protein Data Bank, PDB](https://www.rcsb.org/)— se deriva en gran medida de un mismo tipo de experimentos realizados alrededor del mundo, en los que se hace interactuar a un frágil cristal de alguna proteína, con un haz de alta brillantez de una onda electromagnética ionizante como los rayos X. [1] Pensándolo un poco, es fácil concluir que utilizar esta técnica tiene algunos problemas. Algunos de esos problemas son (1), el PDB es una base de datos redundante (hay mucha información de una misma proteína con cambios sutiles); (2), que se sabe mucho sobre proteínas con forma casi esféricas (globulares, como la albúmina de las claras de huevo y la caseína de la leche) y (3), que son muy afines a estar disueltas en agua (hidrofílicas). Pero el problema principal es que la gran mayoría de la información de tipo tridimensional proviene de experimentos de rayos X, cuya alta energía es por definición, capaz de destruir no sólo la naturaleza ordenada de los cristales de proteína, sino que, a dosis suficientes, pueden matar a organismos vivos.
Recordemos que estas macromoléculas (las proteínas) son cadenas largas, con varios tipos de átomos (C, H, O, N, S y a veces metálicos) y que además se doblan o enrollan (plegamiento) en formas no-azarosas. Así, mientras colectamos datos que darán lugar a una nueva estructura tridimensional, los rayos X destruyen a la proteína, rompiendo enlaces químicos y cambiando la ubicación física de los átomos del cristal (eventos radiolíticos). Este problema asociado con la técnica ha llevado a la comunidad de cristalógrafos de proteínas a desarrollar técnicas de contención del daño que van, desde utilizar temperaturas muy bajas (usualmente a 100 K, o bien, -173.15 °C), hasta la atenuación de la intensidad del haz de rayos X para reducir el efecto ionizante sobre el cristal; es decir esa que afecta los electrones a los átomos, como dijimos, rompiendo enlaces de todo tipo).
Sin embargo, nuestra aproximación como grupo a este problema, es sutilmente diferente: lo primero que hemos intentado es describir: ¿qué es lo que exactamente ocurre dentro de un cristal cuando se ‘acumula’ la radiación de rayos X? Trabajando en laboratorio, analizando publicaciones de otros investigadores y discutiendo intensamente comprendimos que se trataba básicamente, de un proceso de reducción química producida por una cascada de electrones, que se liberan principalmente de las moléculas de agua presentes en el cristal, por efecto de los rayos X. Éstas moléculas ocupan, en promedio, el 45 % del volumen de un cristal de proteínas, asociadas a la superficie de cada proteína, aunque también son cruciales las características fisicoquímicas de cada uno de los aminoácidos que tienen los 20-30 modelos de proteínas con las que trabajamos.
Con esta idea, empezamos entonces a usar esta herramienta metodológica para estudiar procesos inter- e intramoleculares que deben ocurrir en los sistemas celulares de seres tanto extremófilos (que viven a temperaturas, presión o concentraciones muy altas de sal, p. ej., los organismos que viven alrededor de ventilas hidrotermales submarinas [2]) como mesofílicos (viviendo como nosotros los humanos, en condiciones ‘normales’ de temperatura o concentración de oxígeno entre otros factores). Iniciamos experimentos de difracción para ya no solo para hacer fotografías ‘fijas’ de proteínas aparentemente estáticas, sino películas; más bien la conjunción de decenas de fotografía ‘fijas’ que, progresiva y lentamente van mostrando modificaciones causadas por los propios rayos X, de un estado a otro, (partiendo del inicio de la interacción de la enzima con su sustrato, hasta ver a los productos aparecer y salir de la enzima), casi de la misma manera en que lo harían en los sistemas vivos [3].
Actualmente, estamos haciendo este análisis con decenas de proteínas que están implicadas en la reparación del ADN, la degradación de contaminantes ambientales; con enfermedades virales como el SARS-CoV-2, y un largo etcétera que muestra que, un grupo de una veintena de personas, diverso, con intereses amplios y complejos (que en su mayoría son estudiantes de posgrado), los reúne el convencimiento de que el estudio de la estructura tridimensional de las proteínas —tomando en cuenta sus movimientos a distintos niveles, es fundamental para entender una gran cantidad de fenómenos moleculares de la vida [4-7].
Y bueno… para lograr eso, pasamos mucho tiempo buscando maneras en que las proteínas que nos importan lleguen a cristalizar (en sentido literal) [Figs. 3 y 4]. Aparte de eso todo lo que comprendemos a partir de ellos, es una experiencia placentera, casi artística y definitivamente cultural.

Figura 3. Cristales de lisozima de huevo de gallina crecidos por la estudiante de doctorado la M. en C. Angela Escudero García en el grupo de Bioquímica Estructural del IBt.
¿Cuál es el futuro en la biología estructural?
Después de casi tres décadas de existencia como grupo de investigación, es importante de pensar en lo que sigue, y sobre todo en que está pasando en este momento en este campo: estamos frente a un umbral de cambios conceptuales y de retos metodológicos muy importantes para la biología estructural.
Simplemente conocer la estructura tridimensional ‘real’ o más probable de una biomolécula es y seguirá siendo un logro, sobre todo si se trata de proteínas con una forma o un plegamiento desconocido; o bien cuando se trata de proteínas ‘multidominio’ —con varios módulos ordenados uno tras otros como el un collar de perlas— como las que adornan la superficie de virus o bien, proteínas de membrana que participan como receptores, canales iónicos, transportadores y proteínas de amarre, participando en mecanismos de intercambio iónico, transducción de señales, fagocitosis, etc. Sin embargo, muchas de estas están poco representadas en el PDB y como sabemos, ¡el diablo esta siempre en los detalles!, y ahí en el PDB actual y futuro están las pistas de los descubrimientos que, al comprender y modificar proteínas cruciales, pueden seguir modificando cosas tan trascendentes como nuestra calidad o esperanza de vida. Es en estos detalles (sospechas, hipótesis, predicciones, simulaciones e interpretaciones) sobre la forma en que se mueven las proteínas, de donde obtendremos conocimientos sobre procesos tan importantes como la migración celular, el envejecimiento y el control de enfermedades, así como como producir nuevas proteínas que sustituyan materias primas o reduzcan la contaminación que nuestras actividades producen en todo nuestro planeta.
Estamos en el momento en que la biología estructural está dejando de ser solamente un proceso de generación de hipótesis basadas en una aproximación técnica, a una descripción dinámica en un contexto biológico, dando lugar a lo que ya se nombra “Biología estructural integrativa”. En este cambio de paradigma -es decir el modelo ‘en boga’ sobre cómo funcionan ciertas cosas- el concurso de la cristalografía (que permite saber cómo se ven las proteínas en el espacio), con la resonancia magnética nuclear (que permite saber cómo se mueven las proteínas), la crio-microscopía electrónica (que permite observar grupos de proteínas, a 100 K de temperatura), bajo condiciones diferentes a las de un cristal, formando verdaderas ‘fábricas metabólicas’ en las células; la dispersión de rayos X de ángulo bajo y la tomografía de rayos X (que permiten explorar la superficie de las proteínas). Todo esto se complementa y articula con el uso de modelos computacionales provenientes de algoritmos basados en inteligencia artificial, con gran capacidad de simular y predecir condiciones, que serán cada vez más dominantes para el avance del campo. Pero, para que esta y otras áreas de la ciencia desarrolladas en el IBt continúen tan productivas como hasta la fecha, necesitamos dar la oportunidad a que gente joven y apasionada mueva los límites, el nuevo reto es permitir que la pasión que un día encontramos florezca en otros.

Figura 4. Cristales de una proteína fluorescente producidos por el Dr. Víctor Rivelino Juárez González en el IBt.
Lecturas recomendadas y Referencias:
- Rudiño-Piñera E, V Quintero-Hernández y VR Juárez-González (2022). El Protein Data Bank (PDB) y su impacto en la investigación científica. Alianzas y Tendencias BUAP 7(25): 21-35. enlace
- Rodríguez-Arteaga A y J Ramírez-Ramirez (2020). Descifrando el secreto de una proteína resistente a la radiación.Biotec Mov 22: 3-7 .
- Pastor-Colón CN y E Rudiño-Piñera (2020). Danza Molecular. Hypatia, 62. https://www.revistahypatia.org/biologia-estructural-rev-62.html
- De la Mora, E., J. E. Lovett, C. F. Blanford, E. F. Garma, B. Valderrama, E. Rudiño-Piñera (2012). Structural changes caused by radiation-induced reduction and radiolysis: the effect of X-ray absorbed dose in a fungal multicopper oxidase. Acta Crystallogr D Biol Crystallogr 68 (5): 564-577. DOI:10.1107/S0907444912005343
- Serrano-Posada, H., S. Centeno-Leija, S. P. Rojas-Trejo, C. Rodríguez-Almazan, V. Stojanoff, E. Rudiño-Piñera (2015). X-ray-indiced catalytic active-site reduction of a multicopper oxidase: structural insights into the proton-relay mechanism and O2-reduction states. Acta Crystallogr D Biol Crystallogr 71 (12): 2396-2411. DOI:10.1107/S1399004715018714
- Miranda-Blancas, R., M. Avelar, A. Rodriguez-Arteaga, A. Sinicropi, E. Rudiño-Piñera (2021). The b-hairpin from the Thermus thermophilus HB27 laccase works as a pH-dependent switch to regulate laccase activity. J Struct Biol 213 (2): 107740. DOI:10.1016/j.jsb.2021.107740
- Marin-Tovar Y, H Serrano-Posada, A Diaz-Vilchis, E Rudiño-Piñera (2022). PCNA from Thermococcus gammatolerans: a protein involved in chromosomal DNA metabolism intrinsically resistant at high levels of ionizing radiation. Proteins [Struct Funct & Bioinf] 90 (9): 1684-1698. DOI:10.1002/prot.26346
Comparte este artículo en redes sociales

Acerca de los autores
El Dr. Enrique Rudiño Piñera es investigador titular C del Instituto de Biotecnología de la UNAM y responsable del grupo de Bioquímica Estructural del IBt.
Contacto: enrique.rudino@ibt.unam.mx